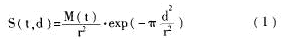论土壤微生物的宏基因组学及其研究进展
来源:岁月联盟
时间:2010-07-11
论文摘要 土壤宏基因组学技术是近来比较迅速的一种新方法,是研究土壤微生物生态学的基础,也是获是土壤中各种基因资源的一种有效手段。介绍了土壤宏基因文库的构建、筛选及土壤宏基因组研究现状和这一技术的局限性。
宏基因组学源于20世纪70年代土壤微生物基因组DNA的直接提取技术的实现,随着微生物学和生物技术的不断发展,Handelsman于1998年提出了宏基因组学的概念[1]。宏基因组学又叫环境基因组学(Environmental Genomics)或群体基因组学(Community Genomics),定义为利用基因组技术直接研究状态环境中的有机体群落,而不需要分离、培养单一种类的微生物。生物学和化学的结合孕育了宏基因组学的诞生,而宏基因组学的发展需要最大限度的利用现代生物技术和实用筛选技术。
土壤宏基因组学技术是近来发展比较迅速的一种新方法[2],这种方法从土壤环境样品中直接提取微生物基因组DNA(宏基因组)并克隆于不同载体,再将重组载体转移到适宜的宿主以建立宏基因组文库;同时结合不同的筛选技术,从基因文库中筛选新基因或新的生物活性物质。应用这些免培养的新方法和新技术,可以绕过微生物菌种分离培养这一技术难关,直接在基因水平上研究、开发和利用无法培养的微生物资源;有利于揭示不可培养微生物的基因多样性,为农业、医药和环境的可持续发展提供丰富的资源。
1土壤宏基因文库的构建
关于土壤宏基因组学技术的构建已有许多研究报道[3],文库的构建需要足够高质量的DNA,由于土壤微生物往往会与土壤其他组分紧密结合,这就增加了提取土壤DNA的难度[4]。常用的方法包括直接提取法和间接提取法。直接提取法是将样品直接悬浮在裂解缓冲液中处理,使其释放DNA,继而抽提纯化;间接提取法是首先去除土壤等杂质,通过不同的离心速度从土壤中分离出细胞,然后对细胞进行抽提。直接提取法提取的DNA片段较小(1~50kb),提取率高,操作简单;间接提取法提取的片断较大(20~500kb),纯度高,但操作繁琐,有些微生物在分离过程中会丢失。
根据插入片断大小,可以把基因文库分成2类:质粒载体的小片段插入(小于15kb)和柯斯质粒(15~40kb)或BAC(细菌人工染色体)(超过40kb)载体的大片段插入。大肠杆菌(Escherichia coli)是表达土壤细菌基因或基因簇的通用宿主,穿梭载体或BAC文库可将大肠杆菌包含的文库信息转移至其他宿主如链霉菌或假单胞菌[5]。
载体系统的选择取决于所提取土壤DNA的质量及研究目的,包括欲插入目的片段的大小、所需要的载体拷贝数、使用的宿主以及筛选方法等。如对腐殖质含量较高或剪切较严重的DNA样品适宜构建质粒文库,小片段的文库适用于筛选新的与代谢相关的单基因或小操纵子;而对于含较大基因簇或大片段的DNA样品则需要构建大片段和大容量的载体文库。Rondon直接把环境DNA克隆到低拷贝BAC载体,以大肠杆菌作为宿主构建了含100Mbp 的小文库(SL1),
并从这个文库中检测到DNA酶、脂肪酶、淀粉水解酶的活性。
2土壤宏基因组文库的筛选
宏基因文库的筛选主要有功能驱动筛选、化合物结构水平的筛选、序列驱动筛选,底物诱导基因表达筛选。功能驱动筛选是根据重组克隆产生的新活性进行筛选,在上有很多重要的酶就是用这种方法发现的。其主要缺点是要在寄主中进行功能表达,造成筛选工作量大,效率低。化合物结构水平的筛选是根据不同结构的物质在色谱中有不同的峰值,通过比较转入和未转入外源基因的宿主细胞或发酵液抽提物的色谱图筛选产生新结构化合物的克隆子。此方法工作量大,费用高。序列驱动筛选是不依赖重组基因在宿主中表达来筛选,而是根据已知功能的基因序列设计探针或PCR引物,通过杂交进行筛选具有目标序列的克隆子。底物诱导基因表达筛选是利用底物诱导克隆子分解代谢基因进行筛选,这种方法已经成功的从宏基因中筛选出芳烃化合物诱导的基因。国内外的资料显示这4种筛选方法可以筛选到所需要的物质,但筛选效率低,费用高。
3土壤宏基因组研究现状
利用宏基因组学的技术,科研人员筛选到了许多功能基因,加拿大TerraGen Discover公司最先在以链霉菌为宿主的宏基因组文库中筛选到了具有抗菌活性的5种新的小分子物质TerragineA、B、C、D、E[5];Courtois等利用柯斯载体构建了含5 000个克隆子的环境基因组文库,采用PCR 序列分析的方法,筛选出编码聚酮合成酶的新基因,同时采用HPLC技术发现了脂肪二烯醇中2种新的化合物,两者互为同分异构体;Yun等选用pUC19为克隆载体构建大肠杆菌基因组文库,利用活性筛选方法,从30 000个重组子中筛选出1个含淀粉酶基因(amyM)的克隆子。
2005年,LimHK等以枯草芽孢杆菌为宿主,建立了森林土壤的宏基因组文库,筛选到2个具有抗菌活性的克隆,对其结构进行分析,得出其中一个为产红色色素的靛玉红,另一个为产蓝色色素的靛蓝,是靛玉红的异构体。2006年,VogetS等首次研究了从土壤宏基因组文库中筛选到的一种纤维素酶的性质,证实了其具有较广的pH值和温度适应范围,并且在较高的盐度时也具有活性,具有工业应用价值。
4土壤宏基因组学的技术局限性
总DNA提取技术尚存在一定的限制,土壤环境中,由于微生物与土壤颗粒紧密结合的特性以及腐殖酸等抑制性物质存在等原因,从中难以获得适于构建宏基因组文库的高分子量DNA。Bertrand等采用间接提取法,通过Nycodenz梯度离心,所回收的土壤DNA片段大小己能达到400kbp,但至今基于原位裂解获得>100kbp土壤DNA的提取技术尚未突破,运用原位裂解法构建更大片段环境宏基因组文库(现有的土壤宏基因组文库中,平均插人片段最大为44.5kb)仍是一个难点。不可避免地,环境宏基因组文库所包含微生物基因组信息的偏差将直接导致“基因遗漏”现象发生,如海洋中普遍存在的微生物固氮基因,却在测序量高达1.6Gbp的马尾藻海水宏基因组文库中被遗漏,表明仅运用宏基因组学技术同样会忽略部分的微生物资源。
阳性克隆筛选频率低是宏基因组学的另一个瓶颈所在,运用经典的功能筛选方式,往往是在数千个,甚至数百万个重组克隆子中才能检测到有用的活性克隆,造成此局面一个重要的原因是外源基因的异源表达水平低下。目前根据核酸序列相似性及基因保守区设计探针或引物的杂交、PCR筛选方法,从文库中发现新基因的比率尚不到已知基因的40%。
环境微生物宏基因组研究能让我们发现存在于不可培养微生物中的一些重要的生理过程。许多实验室已经开始努力从不可培养土壤生物的基因组序列中去获得重要的基因,特别是从Acidobacterium组中,因为它是土壤中最常见的细菌。这些数据提供了关于这些生物在土壤中扮演角色的线索。随着足够序列信息的获得,这些生物的代谢途径将被构建出来,引导我们采取有效的策略去培养这些生物。这些数据也将准许我们构建包含所有已知开放阅读框的微生物芯片,来确定这些基因在时间和空间上的表达状况。
尽管宏基因组技术本身还存在着一些局限性,但它为土壤微生物的研究提供了一种有效的研究策略,尤其是对于99%以上不能获得纯培养的土壤微生物来说,宏基因组学不仅是研究土壤微生物生态学的坚实基础,更是我们获得土壤中各种基因资源的一个有效手段。
5
[1] HANDELSMAN J,RONDON M R,BRADY SF,et al.Molecular biological access to the chemistry of unkown soil microbes:a new frontier for natural products[J].Chen Biol,1998,5(10):245-249.
[2] VOGET S,LEGGEWIE C,UESBCK A,et al.Prospecting for novel bicoatalysts in a soil metagenome[J].Appl.Emiron.Microbiol,2003(69):6235-6242.
[3] RONDON MR,AVGUST PR,BETTENMANN AD,et al.Cloning the soil metagenome:a strategy for accessing the genetic and funcfional diversity of uncultured microorganisms[J].Appl.Environ.Microbiol,2000(66):2541-2547.
[4] SALEH-LAKHA S,MILLER M,CAMPBELL,RG,et al.Microbial gene expression in soil:methods,applications and challenges[J].Micro biological Methods,2005,63(1):1-19.
[5] WANG GY,GRAZIANI E,WATERS B,et al.Novel natual products from soil DNA libraries Tn a streptonycete host[J].Org Lett,2000(2):2401-2404.
下一篇:对我国当前农地改革新路径探析