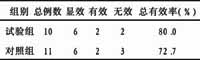
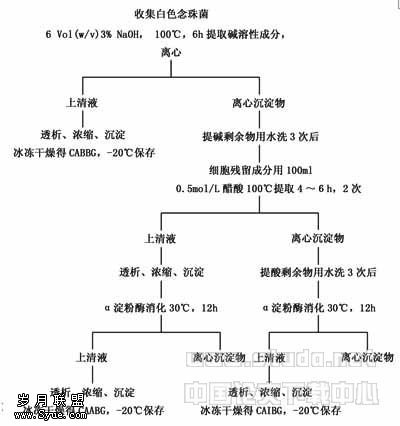
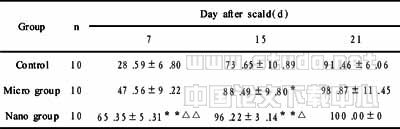
啤酒酵母鲨烯合酶基因的克隆及其基因表达载体的构建
作者:冯丽玲,卢文婕,曾庆平
【摘要】 【目的】 克隆啤酒酵母鲨烯合酶基因及构建其基因表达载体,为在微生物体内重建青蒿素合成途径打下基础。【方法】采用聚合酶链反应(PCR)扩增、扩增片段与T?载体连接和克隆片段的序列分析;酶切并回收目的基因,反向插入酵母表达载体pGAPZαA,双酶切鉴定重组子。【结果】 通过PCR扩增获得1 335 bp片段,克隆后经PCR和酶切反应进行了鉴定。初步测序结果与GenBank上已登录的酵母鲨烯合酶基因序列基本吻合,相差仅数个碱基对;将此片段反向插入酵母表达载体pGAPZαA,构建了反义酵母表达载体,并通过双酶切加以鉴定。【结论】 成功克隆了啤酒酵母鲨烯合酶基因并进行了测序,同时构建了反义酵母表达载体。
【关键词】 啤酒酵母;鲨烯合酶基因;基因复制;反义酵母表达载体
青蒿素是我国自主研发的抗疟原虫中药有效单体,对多药抗性疟原虫有效,现已成为全球抗疟药的希望。若将青蒿素生物合成途径从青蒿“搬到”酵母中,就能在发酵罐内实现青蒿素的化生产。为了探索在酵母内导入鲨烯合酶基因反义序列抑制鲨烯合酶基因表达而使青蒿素合成增加的可能性,本研究克隆了酵母鲨烯合酶基因,并经测序予以证实,同时构建反义酵母表达载体,为下一步培育青蒿素生产酵母菌打下了基础。现报道如下。
1材料与方法
1?1菌种啤酒酵母为INVSc1菌株 购自Invitrogen公司;大肠杆菌JM109 由本实验室保存。
1?2质粒酵母表达载体pGAPZαA购自Invitrogen公司;pMD18?T载体购自Takara公司。
1?3主要试剂与仪器酵母基因组DNA提取试剂盒(离心柱法)、质粒DNA纯化试剂盒(Concert Rapid Plasmid Miniprep System)、DNA片段回收试剂盒(Concert Rapid Gel Extraction System)均 购 自Omega 公司; 聚合酶链反应(PCR)扩增试剂盒 ( One Shot LA PCR Mix)均购自Takara公司; GL?20G?Ⅱ型 高速冷冻离心机(上海); 5332 Mastercycler personal型PCR 仪(德国);BG?subMINI 迷你型水平电泳仪(北京);MiniSpin 5452 个人型高速离心机(德国);260010 MicrocoolerⅡ型制冷仪(美国)。
1?4DNA的提取取5×107酵母细胞加入600 μL山梨醇缓冲液和50 U Lyticase,充分混匀,30 ℃处理30 min,4 000 r/min离心10 min;弃上清液,收集沉淀,加入200 μL裂解液重悬沉淀;加入20 μL蛋白酶K,振荡混匀;加入220 μL缓冲液并充分混匀后,置于70℃水浴15 min;等溶液变清亮,12 000 r/min离心数秒;加入220 μL的无水乙醇,剧烈振荡混匀数秒,此时会出现絮状沉淀;将离心柱装在收集管上,然后将所得溶液及沉淀都转移入离心柱中;12 000 r/min离心1 min,倒掉收集管中的滤液;加入500 μL蛋白清洗液,12 000 r/min离心1 min;倒掉收集管中的滤液;并将离心柱放回收集管中;加入500 μL洗涤液,12 000 r/min离心30 s;倒掉收集管中的滤液,并将离心柱放回收集管中; 重复前一步骤操作1次;12 000 r/min离心1 min,去掉离心柱中的所有液体;将离心柱放在一个干净的、新的1?5 mL离心管中,置于37 ℃烘箱放置5~10 min,待管中的乙醇全部挥发为止; 往离心柱中央悬空滴加经70 ℃水浴预热的100~200 μL洗脱液;室温放置2 min后,12 000 r/min离心30 s,洗脱DNA到离心管中;为了得到更多的DNA,再加100~200 μL洗脱液,室温放置2 min后,再次洗脱DNA。
1?5引物设计按GenBank M63979[1]设计引物SSYPri5:5’?GGTACCATGGGAAAGCTATTAC?3’;
SSYPri3:5’?GAGCTCTCACGCTCTGTGTGTAAAGTG?3’;
YSS?L:5’ ?CCGCGGCCGCAGTGCGAGACACATTTCAC?3’;
YSS?R:5’?GGTCTAGATACCCTTTCGATAATGTTAAC?3’,委托Takara公司合成。
1?6PCR 扩增以提取的酵母DNA为模板,SSYPri 5、SSYPri 3为上下引物,用Takara公司的PCR扩增试剂盒( One Shot LA PCR Mix)扩增,扩增条件为: 94 ℃、1 min→94 ℃、30 s,53 ℃、1 min,72 ℃、3 min,30循环→72 ℃、10 min→4 ℃。
1?7基因克隆将PCR产物与pMD18?T载体连接,转化JM109感受态细胞,涂板,蓝白斑筛选重组子。
1?8重组质粒鉴定及测序用质粒DNA纯化试剂盒提取质粒,分别经PCR扩增和HimIII、KpnI酶切进行鉴定 。PCR扩增采用Takara公司的Tf1酶,反应条件为:94 ℃、1 min→94 ℃、30 s,57℃、1 min,72 ℃、5 min,30循环→72 ℃、10 min→4 ℃。最后在 ABI Prism 310 DNA测序仪上用RV?M引物测序。
1?9表达载体的构建因GenBank上已登陆的酵母鲨烯合酶DNA序列显示无内含子存在,即酵母鲨烯合酶DNA序列与cDNA序列相同。因此本研究设计以pT?YSS重组质粒为模板,利用自定义YSS?L 和YSS?R扩增引物进行PCR扩增,扩增条件为:94 ℃、1 min→94 ℃、30 s,57 ℃、1 min,72 ℃、5 min,30循环→72 ℃、10 min→4 ℃。将NotI和XbaI酶切PCR产物后所得片段插入相同酶切的pGAPZαA载体中,获得中间表达载体pGAPZ?YSS。以pGAPZ?YSS转化JM109感受态细胞,挑取单菌落,用NotI和XbaI进行双酶切鉴定。
2结果
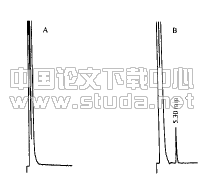
2?1鲨烯合酶基因扩增酵母DNA经PCR扩增和10 g/L凝胶电泳后,可检测到长度约为1 335 bp的DNA区带(图1)。
2?2鲨烯合酶基因克隆将扩增片段插入pMD18?T载体后获得重组质粒pT?YSS。对pT?YSS进行1. 100 bp ladder;2~4. PCR产物
图1酵母DNA的PCR扩增产物
Figure 1Results of PCR amplification of yeast DNAPCR扩增,经10 g/L凝胶电泳后检测到与酵母DNA的PCR产物大小相当的区带,表明上述扩增片段已与载体连接。将重组质粒用HindIII和KpnI酶切,经10 g/L凝胶电泳后,可见一条略比酵母DNA的PCR产物大的基因区带和另一条载体带(图2)。
按实验设计,鲨烯合酶基因扩增片段的理论长度应为1 335 bp,插入T载体并用HindIII和KpnI双切后,含有鲨烯合酶基因的酶切片段中尚包括45 bp载体片段,其实际长度应为1 380 bp。实验结果与此分析相符,初步表明酵母青蒿鲨烯合酶基因已被克隆。
2?3鲨烯合酶基因的序列分析对上述重组质粒中的插入片段进行序列分析,结果显示pT?YSS的插入片段为1 335 bp。经与GenBank中所发表的序列比较,显示两者序列基本吻合,但序列之间存在某些碱基对的差别,推测这些差异可能属于单核苷酸多态性(SNP)。本实验克隆的鲨烯合酶基因已被GenBank收录,编号为EU675328。 测序结果与GenBank上已登陆的酵母鲨烯合酶基因序列同源性为99%,氨基酸序列同源性为100%。
2?4反义鲨烯合酶基因载体鉴定按预先设计的反义YSS基因表达载体构建方案,将携带NotI和XbaI酶切位点的YSS基因PCR扩增产物与pGAPZαA同时用NotI和XbaI消化,然后经T4 DNA连接酶连接而获得重组质粒pGAPZ?YSS(图3)。
为了确保反义YSS 基因序列准确无误,还对pGAPZ?YSS中的YSS插入片段进行了重测序,结果表明序列完全正确,且排列方向相反。
3讨论
青蒿素是我国自主开发的强效、低毒、无抗药性抗疟药,尤其是作为脑型疟疾和抗氯喹恶性疟疾的特效药,被世界卫生组织(WHO)誉为“治疗疟疾的最大希望” [2]。但天然青蒿中青蒿素含量极低,难以满足抗疟药市场需求。为此,全球竞相开展了在微生物体内重建青蒿素合成途径的研究[3-5]。
在青蒿素合成代谢中,从乙酰辅酶A至法尼基焦磷酸(FPP)的8个酶促步骤共存于青蒿及酵母中,而紫穗槐?4,11?二烯合酶(ADS)催化的FPP环化反应为青蒿所特有[6]。随后紫穗槐?4,11?二烯(AD)→青蒿醇12→青蒿醛→双氢青蒿醛→双氢青蒿酸为细胞色素P450(Cyt P450)酶系催化的酶促反应[7-8],双氢青蒿酸→双氢青蒿酸过氧化物→青蒿素为自由基介导的非酶促反应[9-10]。由于酶促反应过程复杂,非酶促反应条件苛刻,目前国内外尚未重建青蒿素合成途径。我们拟尝试:(1)开源:将青蒿素合成关键酶ADS基因导入酵母,以获取青蒿素前体AD;(2)节流:抑制酵母中麦角固醇合成的关键酶——SS基因,以使碳源更多地流向AD;(3)将AD用青蒿酶提取液进行生物转化,为青蒿素大规模化生产作准备。
因此,为了达到节流目的及研究在酵母内导入鲨烯合酶基因反义序列抑制鲨烯合酶基因表达使青蒿素合成增加的可能性,本研究首先克隆了酵母鲨烯合酶基因,并经测序予以证实,同时构建反义酵母表达载体,然后在这一工作的基础上,拟将反义SS基因整合到酵母染色体上,抑制酵母中麦角固醇合成的关键酶——SS基因,使甲羟戊酸途径中的碳源更多地引到AD中来(节流),从而为下一步培育青蒿素生产菌打下了基础。
【】
[1] Jennings S M, Tsay Y H, Fisch T M,et al. Yeast squalene synthetase gene, complete cds[EB/OL].GenBank M63979.
[2] WHO. Antimalarial drug combination therapy,report of a WHO technical consultation[C]. April 2001.
[3] Ro D K, Paradise E M, Ouellet M, et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast[J]. Nature, 2006, 440: 940.
[4] Martin V J, Pitera D J, Withers S T, et al. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids [J]. Nat Biotechnol, 2003, 21: 796.
[5] Lindahl A L, Olsson M E, Mercke P, et al. Production of the artemisinin precursor amorpha?4,11?diene by engineered Saccharomyces cerevisiae [J]. Biotechnol Lett, 2006, 28: 571.
[6] Bouwmeester H J, Wallart T E, Janssen M H A, et al. Amorpha?4,11?diene synthase catalyse the first probable step in artemisinin biosynthesis. [J]. Phytochemistry, 1999, 52: 843.
[7] Mercke P, Bengtsson M, Bouwmeester H J, et al. Molecular cloning, expression, and characterization of amorpha?4,11?diene synthase, a key enzyme of artemisinin biosynthesis in Artemisia annua L[J]. Arch Biochem Biophys, 2000, 381: 173.
[8] Chang Y J, Song S H, Park S H, et al. Amorpha?4,11?diene synthase of Artemisia annua: cDNA isolation and bacterial expression of a terpene synthase involved in artemisinin biosynthesis [J]. Arch Biochem Biophys, 2000, 383: 178.
[9] Wallaart T E, Van Uden W, Lubbeerink H G M, et al. Isolation and identification of dihydroartemisinic acid from Artemisia annua and its role in the biosynthesis of artemisinin[J]. J Nat Prod, 1999,62:430.
[10] Wallaart T E, Pras N, Quax W J. Isolation and identification of dihydroartemisinic acid hydroperoxide from Artemisia annua: a novel biosynthetic precursor of artemisinin[J].J Nat Prod, 1999, 62: 1161