血安片质量标准研究
作者:冮海峰 胡跃兰 孟祥军
【摘要】 目的:按新药审批标准为血安片制定质量标准。方法:以TLC法对血安片中棕榈子进行定性鉴别;以HPLC法测定血安片中没食子酸的含量。结果:试验采用薄层色谱法定性鉴别,色谱斑点清晰,分离度良好,阴性液无干扰;以HPLC法测定该片剂中没食子酸的含量,方法精密度、稳定性、重复性良好,回收率为97.28~103.22%, RSD为2.0%(n=9)。结论:本试验所确定的质量分析方法稳定可靠,能作为本片剂的质量控制标准。
【关键词】 血安片 质量标准 方法学研究
【Abstract】Object:formulate the quality standard for the Xue,an tablets according to the requirement by Evaluation of Chinese Government。Methods:the qualitative identifying method by TLC was established for Zonglvzi and gallic acid was quantitatively determined by HPLC。Result shows:the spots on the TLC are clear by the methods of qualitative identification by TLC,the resolution is well and have no negative sample interference。The method for determinating the content of tablets by HPLC is suitable for the formulate of the quality standard with sufficient accuracy、stability and reappearance 。The recovery rate is 97.28~ 103.22 %,RSD is 1.92% (n=9)。Conclusion:the qualitative and quantitative analytic methods are stable and reliable。
【Keyword】The Xue,an tablets Quality Standard Method Appraise
血安片是根据已有部颁标准的血安胶囊[1]改变剂型得到的中成药制剂。是由棕榈科植物棕榈子的干燥成熟果实经乙醇提取加工制成的片剂,属中药八类新药。本品应用于临床上具有止血、收敛、调经之功效,用于月事不准、经血过量、崩漏、淋漓不止、产后恶露不尽等妇科出血症。为保证临床用药安全有效,严格控制此片剂的质量,特对处方中棕榈子[2]的定性、定量方法加以筛选和分析,从而确立稳定可靠的质量标准。
原血安胶囊标准[1]鉴别项下①为显色反应,②为TLC薄层鉴别。通过实验证明,鉴别①现象明显,具有重现性,可以继续沿用。而对鉴别项②进行试验的结果表明,此标准鉴别项下的薄层条件未能使棕榈子中的原儿茶酸达到有效的分离。通过查阅相关资料,我们采用“复方丹参口服液中药质量标准”[3]中的鉴别项①的展开剂条件对本品的鉴别项进行了试验,试验结果证明此展开剂能够满足原儿茶酸的分离要求,且棕榈子中的原儿茶醛在此条件下也能显色,但与其他组分的分离效果并不理想,所以我们对此展开剂进行了优化,将展开剂苯-醋酸乙酯-甲酸(80︰50︰4)的比例更改为(70︰40︰4),此条件可以使原儿茶酸、原儿茶醛同其他组分有效的分离,所以特增加了棕榈子中原儿茶醛的鉴别,显色现象明显。经上述试验,修订了棕榈子的薄层鉴别。
我们参照文献“复方珍珠口疮颗粒中药质量标准”[4]中没食子酸含量测定法对本品中所含的没食子酸进行测定。试验结果表明,没食子酸峰与其他杂质峰能够有效分离,其方法能够满足控制本品含量的要求,故我们采用文献中的HPLC法,建立了高效液相色谱条件,测定制剂中没食子酸的含量,其方法简便、准确、专属性强。同时考虑到本品中没食子酸含量,我们将对照品与供试品的取样量进行了适当的调整。
1 实验资料
1.1 处方 棕榈子10000g。
1.2 制法 将棕榈子粉碎成粗粉,用5倍量的95%乙醇浸泡20分钟,加热提取2小时,回收乙醇,滤过,滤液浓缩至相对密度1.20~1.22(60℃)的清膏,干燥,粉碎成细粉,加入适量的辅料,混匀,制成颗粒,干燥,压制成素片1000片,包薄膜衣,即得。
2 性状
本品为暗红色薄膜衣片;除去包衣后显棕红色,味涩。
3 薄层定性鉴别
本制剂是由棕榈子经乙醇提取加工制成的薄膜衣片,片重0.55g,其中含原药粉0.5g,相当于原药材10g。除去包衣后显棕红色,味涩。本品化学成分明确,对具有的对照品重点进行了筛选,最后确立采用TLC法对棕榈子中原儿茶酸、原儿茶醛进行定性鉴别,试验条件与方法说明如下。
3.1 展开剂的选择
本实验先后采用氯仿-丙酮-甲醇-冰醋酸(7︰1︰1︰0.5)[2](Ⅰ),氯仿-甲醇-水(13︰7︰1)[5](Ⅱ),氯仿-丙酮-甲醇-冰醋酸(7︰2︰0.5︰0.5)[6] (Ⅲ),苯-醋酸乙酯-甲酸(80︰50︰4)[3](Ⅳ)为展开剂,在系统(Ⅳ)的条件下,供试溶液中原儿茶酸与相邻组分分离度良好,并分离出了原儿茶醛,但分离效果不明显。为了更好的达到分离的目的,对其展开剂比例进行了一定的调整,经过试验验证,苯-醋酸乙酯-甲酸(70︰40︰4)的比例效果最优。本试验曾对苯进行了环己烷、正己烷等化学结构相似的试剂的替代试验,但实验效果不佳,故确立苯-醋酸乙酯-甲酸(70︰40︰4)为展开剂,鉴别其成分原儿茶酸、原儿茶醛。
3.2 棕榈子的TLC法鉴别
仪器:薄层层析装置、烘箱。试剂:所用试剂为分析纯,薄层层析用硅胶G由青岛海洋化工有限公司制造,为化学纯。薄层板:取硅胶G,按1︰3比例加入水,铺成0.3mm厚的薄层板,晾干,105℃活化30分钟,干燥箱内保存。
本试验根据复方丹参口服溶液中药质量标准[标准号:WS-150(Z-30)-92]规定的方法检查方中棕榈子所含的原儿茶酸、原儿茶醛,经采用萃取法去除了部分干扰组分的影响,经多次试验,供试品色谱中,具有与对照品相同位置的相同颜色的斑点,待测组分斑点清晰,方法重复性良好,阴性液无干扰。
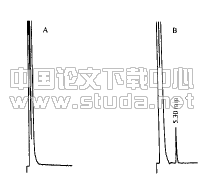
供试品及标准品的制备按照血安片标准,考虑到原胶囊内容物中未添加辅料,故标准应根据药物原粉与辅料的比例适当增加供试品的取样量。具体操作方法:取本品12片,研细,取约5.5g,加水50ml,热浸2小时,滤过,滤液置分液漏斗中,用乙醚提取二次,每次20ml,合并乙醚层,蒸干,残渣加甲醇2ml使溶解,作为供试品溶液。另取原儿茶酸、原儿茶醛对照品,分别加甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(药典2005年版一部附录Ⅵ B)试验,吸取上述三种溶液各20βl,分别点于同一硅胶G薄层板上,以苯-乙酸乙酯-甲酸(70︰40︰4)为展开剂,展开,取出,晾干,喷以2%三氯化铁乙醇溶液。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点,斑点久置加深。
4 检查
照中国药典2005年版一部(附录ⅠD)[7]检查。
4.1 重量差异 照中国药典2005年版一部附录ⅠD的重量差异项下测定,重量差异限度应在标示装量的±5.0%以内,三批样品均符合规定,结果见表1。 表1 重量差异的测定结果结果表明,三批样品重量差异均符合规定。
4.2 崩解时限 照崩解时限检查法(中国药典2000年版一部附录Ⅻ A)测定样品,三批样品(20060304、20060305、20060306)的检查结果见表2。表2 崩解时限的检查结果结果表明,三批样品崩解时限均符合规定。
4.3 微生物限度 照微生物限度检查法(中国药典2005年版一部附录ⅩⅢ C),细菌数限度为100个/g,霉菌、酵母菌数限度为100个/g,大肠杆菌、活螨不得检出。三批供试品(20060304、20060305、20060306)的检验结果,测定结果见表3。表3 微生物限度检查结果结果表明,三批样品微生物限度检查均符合规定。
4.4 砷盐 按砷盐检查法(中国药典2005年版一部附录Ⅸ F)试验。
精密量取每1ml含1μg的标准砷溶液1.0ml、2.0ml、3.0ml,置坩埚中,同供试品砷斑制备方法,依法制备标准确砷斑(依次为A、B、C);取本品10片,研细,取约1g,精密称定,置坩埚中,缓缓灼烧至完全炭化,放冷,加硫酸1ml使湿润,用低温加热至硫酸除尽后,加硝酸0.5ml,蒸干,至氧化氮蒸气除尽,放冷,在540℃灼烧使完全炭化,放冷,加水25ml,定量转移至A瓶中,加碘化钾试液5ml与酸性氯化亚硒试液5滴,在室温放置10分钟后,加锌片2g,立即连接导气管C与A瓶,置37℃水浴中,反应45分钟,取出溴化汞试纸,即得。依法测定三批样品(20060304、20060305、20060306),砷盐含量均低于2ppm,测定结果见表4。表4 砷盐检查结果试验结论:通过对三批样品的检验其砷盐含量均低于2ppm,因此暂不将其列入标准。
4.5 炽灼残渣 按炽灼残渣检查法(中国药典2005年版一部附录Ⅸ J)测定。取本品10片,研细,取约1g,置已炽灼至恒重的坩埚中,精密称定,缓缓炽灼至完全炭化,放冷至室温,加硫酸1ml,使湿润,低温加热至硫酸蒸汽除尽后,在540℃炽灼使完全灰化,移置干燥器内,放冷至室温,精密称定后再在540℃炽灼至恒重。测定三批供试品(20060304、20060305、20060306),炽灼残渣含量均低于0.2%,测定结果见表5。表5 炽灼残渣的测定结果试验结论:通过对三批样品的检验炽灼残渣含量均低于0.2%,因此暂不将其列入标准。
4.6 重金属 按重金属检查法(中国药典2005年版一部附录Ⅸ E第二法)测定。取炽灼残渣项下残渣,分别加硝酸0.5ml,蒸干,至氧化氮蒸汽除尽,放冷,加盐酸2ml,置水浴上蒸干后加水15ml,滴加氨试液至对酚酞指示液显中性,再加醋酸盐缓冲液(PH=3.5)2ml,微热使溶解,移置纳氏比色管中,加水稀释成25ml(乙管:1、2、3);另取配制供试品溶液的试剂,共四份,分别置瓷皿中蒸干,各加醋酸盐缓冲液(PH=3.5)2ml与水15ml,微热使溶解,分别移置纳氏比色管中,分别加每1ml相当于10μg的标准铅溶液0.5ml、1.0ml、1.5ml、2.0ml,加水稀释成25ml(甲管:1、2、3与4)。在甲乙两组管中,加硫代乙酰胺试液各2ml,摇匀,放置2分钟,同置白纸上,自上而下透视,比较甲管与乙管的颜色。结果:乙管1、2、3的颜色均比甲管2的颜色浅。三批供试品(20060304、20060305、20060306)中重金属含量均低于10ppm,测定结果见表6。表6 重金属测定结果试验结论:通过对三批样品的检验重金属含量均低于10ppm,因此暂不将其列入标准。
5 含量测定
本制剂由棕榈子经提取加工制成,经有关文献表明棕榈子中含有原儿茶酸、原儿茶醛、对羟基苯甲酸、d-儿茶素、没食子酸等。药实验表明,没食子酸为棕榈子中主要的止血成分,具有抗凝血和血栓形成等生理活性,故本试验拟HPLC法测定没食子酸的含量,对控制本品的内在质量很有意义。在选定的色谱条件下,阴性液无干扰。经方法学考察,方法的线性、精密度、重现性、稳定性、最低检测限、定量限、阴性对照溶液试验、加样回收试验、样品的含量测定均符合有关规定。因此,确立本制剂定量指标为没食子酸,定量方法为液相色谱法。
5.1 仪器试药
仪器:高效液相色谱仪(UV200 Ⅱ大连依利特仪器有限公司);SunRun Express色谱数据工作站;天平(AL204 梅特勒-托利多上海称重设备公司);色谱柱:Diamonsil C18柱(205×4.6mm,5μm);试药:甲醇-0.1%磷酸水溶液(15︰85),甲醇为色谱纯,其余试剂为分析纯,没食子酸(供含量测定用,110831-200302),中国药品生物制品检定所。
5.2 色谱条件及系统适用性试验考察
5.2.1 测定波长的确立 精密称取没食子酸对照品适量,加20%甲醇制成每1ml中约含12μg的溶液,摇匀,照紫外分光光度法(药典2005年版一部附录Ⅴ A),在200~500nm处扫描,溶液在210、270nm波长处都有最大吸收,但210nm为敏感波长,最后根据[4]确定检测波长为273nm。
5.2.2 流动相的筛选与确立 本实验先后采用甲醇-水-二甲基甲酰胺-冰醋酸(1︰76︰20︰3)[8](Ⅰ),甲醇-0.5%乙酸(15︰85)[9](Ⅱ),甲醇-水-冰醋酸(32︰96︰0.25)[10](Ⅲ),甲醇-水(15︰85)(水相含0.1%磷酸)(Ⅳ),甲醇-四氢呋喃-冰醋酸-水(0.08︰0.64︰2︰97.32)[11] (Ⅴ)为流动相,在系统(Ⅳ)的条件下,供试溶液中没食子酸与相邻组分分离度良好,阴性液无干扰。故确立甲醇-水(15︰85)(水相含0.1%磷酸)为流动相系统。
5.2.3 色谱柱最低理论塔板数考察 色谱柱:十八烷基硅烷键合硅胶;柱温:室温;流动相:甲醇-水(15︰85)(水相含0.1%磷酸);流速:1.0ml/min;检测波长:273nm;测定时间:25min。在此条件下,供试液中没食子酸与其它组分分离度良好,使用的色谱柱为:Diamonsil C18柱(250×4.6mm,5μm)。根据测定结果,确定本实验理论板数按没食子酸色谱峰计不得低于2000。
5.3 线性范围考察
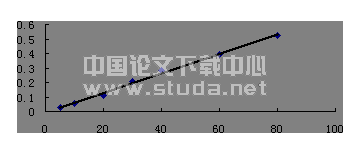
精密称取没食子酸对照品12mg置100ml量瓶中,加20%甲醇使溶解并稀释至刻度,摇匀。再分别精密量取0.8、0.9、1.0、1.1、1.2ml,置10ml量瓶中,加20%甲醇稀释至刻度,摇匀,制成每1ml分别含9.6μg、10.8μg 、12μg 、13.2μg 、14.4μg没食子酸的溶液。精密量取上述溶液各20μl,分别注入液相色谱仪,测定峰面积,结果见表8。以峰面积(Y)对进样浓度(Ⅹ,μg/ml)进行线性回归,回归方程为Y=99483x+17969,相关系数R2=0.9997。结果表明,注入液相色谱中的没食子酸质量在0.192~0.288μg范围内,峰面积与没食子酸浓度线性关系良好。分别精密吸取上述溶液各20μl,注入液相色谱仪中,测定色谱峰面积,测定结果见表7。表7 线性范围考察结果以注入液相色谱中的没食子酸的量为横坐标,色谱峰面积为纵坐标,绘制标准曲线,经线性回归,回归方程为:Y=5E+06x+17969,R2=0.9997,由测定结果提示,注入液相色谱中的没食子酸质量在0.192~0.288μg范围内线性关系良好。
5.4 阴性干扰试验
5.4.1 空白溶液制备 由于本品中只有棕榈子一味药材,故做辅料空白试验,取处方量辅料,按样品制备方法制成阴性液。
5.4.2 供试品溶液的制备 取本品5片,去包衣后研细,取细粉约1.3g,精密称定,置具塞锥形瓶中,精密加甲醇15ml,称重,超声处理(功率 200W,频率先40DHz)30分钟,取出,放冷,称重(补足原重量),摇匀,滤过,滤液经微孔滤膜(0.45μm)滤过,溶液置棕色瓶中备用,即得。
5.4.3 对照品溶液制备 精密称取没食子酸对照品适量,精密称定,加20%甲醇溶解,制成每1ml含12μg的溶液,作为对照品溶液,置棕色瓶中备用,即得。
5.4.4 测定法 精密吸取供试品溶液、阴性液、对照品溶液各20μl,注入液相色谱仪中,记录色谱图,由色谱图提示,在对照品色谱相应的位置上,供试品溶液具有相同保留时间的色谱峰,阴性液在此保留时间处无色谱峰,测定结果提示,其它组分对本品中没食子酸含量测定无干扰,方法专属性良好。
5.5 供试品溶液制备方法考察
鉴于本制剂中棕榈子已经提取后加入,故本试验拟以超声处理法制备供试液。试验中对提取溶剂、超声处理条件进行了定量考察,考察结果如下。
5.5.1 提取溶剂选择 根据没食子酸易溶于水、醇、醚、丙酮的化学性质,并有关文献,本试验拟以20%甲醇、50%甲醇为溶媒,以没食子酸提取量为考察指标。考察方法为:取本品三份,除去包衣,研细,精密称取1.3g,置具塞锥形瓶中,各分别精密加入20%甲醇、50%甲醇和甲醇各15ml,称重,超声处理(功率200W,频率40KHz)30分钟,取出,放冷,称重(补足原重量),摇匀,滤过,滤液经微孔滤膜(0.45μm)滤过,溶液置棕色瓶中备用。分别精密吸取供试品溶液与对照液各20μl,注入液相色谱仪中,按前述液相色谱条件进行分析,结果见表8。表8 提取溶剂考察结果根据测定结果,选用甲醇为提取溶剂,没食子酸含量较高,故确立甲醇为供试品的提取溶剂。
5.5.2 超声处理时间考察 考察方法为:取本品三份,除去包衣,研细,精密称取1.3g,置具塞锥形瓶中,各分别精密加入甲醇15ml,称重,分别超声处理(功率200W,频率40KHz)10、30、60分钟,取出,放冷,称重(补足原重量),摇匀,滤过,滤液经微孔滤膜(0.45μm)滤过,溶液置棕色瓶中备用。分别精密吸取供试品溶液与对照品液各20μl,注入液相色谱仪中,按前述液相色谱条件进行分析,考察结果见表9。表9 超声处理时间考察结果相关资料证明[12],超声提取1小时没食子酸提取完全,根据测定结果超声处理1小时,样品中没食子酸含量最高。但超声处理30分钟提取的没食子酸含量为1小时提取量的99.84%,已基本提取完全。采用30分钟提取样品进行实验,不影响没食子酸含量的测定,故确立以超声处理30分钟为提取方法。
5.6 精密度试验考察
5.6.1 重复性试验考察 取本品5片(批号20060304),去包衣后研细,取细粉约1.3g,精密称定,按正文[含量测定]项下方法制备供试液。分别精密吸取20μl,连续进样6次,按前述色谱条件进行分析,测定色谱峰面积,测定结果见表10。表10 重复性试验测定结果测定结果表明,本试验重复性试验良好,符合有关规定。
5.6.2 重现性试验考察 取六份(批号20060304)样品,去包衣后研细,每份各取细粉约1.3g,精密称定,分别按样品[含量测定]项下方法操作,测定,测定结果见表11。表11 重现性试验测定结果测定结果表明,本试验重现性试验良好,符合有关规定。
5.7 稳定性试验考察
取本品5片(批号20060304)去包衣后研细,取细粉约1.3g,精密称定,按正文[含量测定]项下方法制备供试液。精密量取同一供试液(批号20060304)20μl,按0、2、4、6、8小时时间间隔,分别进样分析,测定色谱峰面积,测定结果见表12。 表12 稳定性试验考察结果试验结果提示,供试液制备后8小时内测定,色谱峰面积无明显变化,在此时间内待测组分化学性质稳定。
5.8 最低检测限
取对照品溶液1ml,置100ml量瓶中,加20%甲醇稀释至刻度,摇匀,再精密量取20μl注入液相色谱仪中,记录色谱图,按3倍信噪比S/N计,最低检测限为2.4ng。
5.9 定量限
取对照品溶液3ml,置100ml量瓶中,加20%甲醇稀释至刻度,摇匀,再精密量取20μl注入液相色谱仪中,记录色谱图。色谱图中,信噪比约为10︰1,定量限约为7.2ng。
5.10 加样回收试验考察
取没食子酸对照品适量,精密称定,加甲醇制成每1ml含12μg的溶液,即得,作为对照品溶液。取本品5片,去包衣后研细,取细粉约1.3g,精密称定,置具塞锥形瓶中,精密加甲醇15ml,称重,超声处理(功率200W,频率40KHz)30分钟,取出,放冷,称重(补足原重量),摇匀,滤过,滤液经微孔滤膜(0.45μm)滤过,溶液置棕色瓶中备用,即得,作为供试品溶液。精密量取供试品溶液与对照品溶液各20μl,分别注入液相色谱仪,测定,测得供试品中没食子酸的含量(C)为140.8μg/g。取没食子酸12mg,置100ml容量瓶中,加20%甲醇使溶解并稀释至刻度,摇匀;再精密量取10ml,置100ml容量瓶中,加20%甲醇使溶解并稀释至刻度,摇匀,制成每1ml含没食子酸12μg,作为加样用对照品溶液。取上述已经测定含量的供试品药粉,分别精密称定约0.9g、1.3g、1.7g各三份,精密称定,置具塞锥形瓶中,精密加甲醇、加样用对照品溶液各15ml,称重,超声处理(功率200W,频率40KHz)30分钟,取出,放冷,称重(补足原重量),摇匀,滤过,滤液经微孔滤膜(0.45μm)滤过,溶液置棕色瓶中备用,即得,作为加样用供试品溶液。精密量取上述各溶液20μl,注入液相色谱仪,记录色谱图,测定含量,计算加样回收率,测得9次平均回收率为99.80%,RSD为2.0%,结果见表13。测得量(mg)=A供/A对×C对×30(ml),样品中没食子酸含量(μg)=供试品取样量×140.8(μg/g)。表13 加样回收试验测定结果测定结果提示:本方法回收率在97.28~103.22%之间,RSD为2.0%,符合有关规定。
根据上述试验结果,制定血安片的含量测定方法如下。①色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.1%磷酸水溶液(15︰85)为流动相,检测波长为273nm。理论板数按没食子酸峰计算应不低于2000。②对照品溶液的制备:取没食子酸对照品适量,精密称定,加20%甲醇制成每1ml含12μg的溶液,即得。③供试品溶液的制备:取本品5片,去包衣后研细,取细粉约1.3g,精密称定,置具塞锥形瓶中,精密加甲醇15ml,称重,超声处理(功率200W,频率40KHz)30分钟,取出,放冷,称重(补足原重量),摇匀,滤过,滤液经微孔滤膜(0.45μm)滤过,溶液置棕色瓶中备用,即得。④测定法:分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,即得。
5.11 含量限度确定的试验
为制定含量限度标准,制备供试品十批(20060301、20060302、20060303、20060304、20060305、20060306、20060307、20060308、20060309、20060310),并与血安胶囊市售品(批号20060227)比较。按上述测定方法,测定样品中没食子酸的含量,结果见表14。表14 样品的测定结果本品的原药材棕榈子收载于卫生部药品标准中药材第一册(1992年版),其中没有对其有效成分没食子酸进行限量,而且查阅相关文献仍没有明确的规定。因此根据上述测定结果,考虑到药材的来源和质量差异对棕榈子回收率的影响,最后将其含量限度暂定为:本品每片含棕榈子以没食子酸(C7H6O5)计,不得少于50μg。
6 讨论
6.1 本试验采用TLC法对方中棕榈子的鉴别方法加以筛选研究,在与对照品色谱相应位置上,供试品色谱斑点清晰,分离度良好,从而确立了重复性、专属性良好的分析方法,其鉴别方法简便、可行。因此确立本制剂定性指标为原儿茶酸、原儿茶醛,定性鉴别方法为TLC法。
6.2 试验中对提取溶剂、超声处理条件进行了考察。结果表明,选用20%甲醇为提取溶剂,超声提取30分钟,样品中没食子酸提取效率最佳。
6.3 复方珍珠口疮颗粒中药质量标准中规定了对没食子酸的含量测定方法与限度,经查阅有关文献,棕榈子中所含没食子酸含量稳定,且没食子酸为棕榈子中主要的止血成分,具有抗凝血和血栓形成等生理活性,故本试验拟HPLC法测定棕榈子中没食子酸的含量。在选定的色谱条件下,供试溶液中棕榈子与相邻组分分离度良好,阴性液无干扰。经方法学考察,方法的重复性、稳定性、精密度、回收率试验均符合有关规定。因此,确立本制剂定量指标为没食子酸,定量方法为液相色谱法。
通过试验,我们对血安片的定性鉴别、含量测定及片剂项下所规定的各项指标进行优化、选择,所确定的质量分析方法稳定可靠,可作为本片剂的质量控制标准。
【参考文献】
[1] 血安胶囊药品标准[S].标准号:WS3-B-2879-98.
[2] 棕榈子[M].卫生部药品标准中药材第一册,1992,89.
[3] 复方丹参口服溶液中药质量标准[S].标准号:WS-150(Z-30)-92.
[4] 复方珍珠口疮颗粒中药质量标准[S].标准号:WS2-091(Z-015)-2001.
[5] 沈海葆,姜华,任志华.棕榈不同制品的薄层层析及止血作用的比较[N].南京中医药大学学报,1995,11(5).
[6] 中药脱牙敏糊剂标准[S].
[7] 中国药典[M].2005年版一部.
[8] 孙立立.HPLC法测定棕榈饮片主要化学成分的含量[J].中药材,1994,9,17(9):28~29.
[9] 董威严.高效液相色谱法测定菱角中的没食子酸含量[J].食品,2005,26(8):334~335.
[10] 胡晓炜,赵青威.RP-HPLC法测定风寒是感冒宁冲剂中原儿茶酸、原儿茶醛的含量[J].中国应用药学杂志,2002,2,19(1).
[11] 苗爱东,王本富,高晓黎.反相HPLC法测定妇康栓中没食子酸的含量[J].西北药学杂志,2000,12,15(6).
[12] 任源.HPLC测定没食子酸的含量[J].华西药学杂志,2004,6,20:71~72.