抗敏颗粒质量标准研究
【摘要】 目的:建立抗敏颗粒剂的质量标准。方法:采用薄层色谱法对抗敏颗粒剂中的当归、桂枝进行薄层色谱鉴别;采用高效液相色谱法对黄芩中的黄芩苷的含量进行测定。结果显示:试验采用薄层色谱法定性鉴别,色谱斑点清晰,分离度良好,阴性液无干扰;以HPLC法测定本品黄芩中的黄芩苷的含量,方法精密度、稳定性、重复性良好,回收率在98.1~100.6%之间,RSD为1.5%。结论:本试验所确定的质量分析方法稳定可靠,能较为合理地对处方中各组分进行定性、定量检测,重复性强,可作为抗敏颗粒的质量标准。
【关键词】 抗敏颗粒剂 质量标准 薄层鉴别 高效液相色谱法
Study on Quality Standards of Kangmin Granule
【Abstract】Object:To establish the quality control method for Kangmin granule.Methods:Radix angelicae sinensis and Ramulus cinnamomi was identified by TLC;and the content of Baicalin were determined by HPLC.Result shows:the spots on the TLC are clear,The determination of Baicalin showed a good linear relationship at arrange of from 0.0944 to 0.9440μg,the average recoveries of Baicalin was from 98.1 to 100.6%,RSD was 1.50%.Conclusion: the qualitative and quantitative analytic methods are stable,reliable and easy to repeat.It can be applied as the standard of Kangmin Granule.
【Key words】Kangmin granule Quality Standards TLC HPLC
抗敏颗粒剂是本单位科研课题,由黄芩、当归、桂枝等药组成。具有补气固表、清热、养血之功效,临床用于变态反应性疾病获得了较好的疗效。本文对当归、桂枝进行了定性鉴别,并以高效液相色谱法测定黄芩中黄芩苷的含量。结果显示,方法可靠,重复性强,可作为质量控制的定量指标[1]。
1 仪器与试剂
1.1 仪器 安捷伦(LC1100)液相色谱仪;G21771AA-UV检测器;安捷伦色谱工作站;KQ3200超声波清洗器(江苏昆山)。
1.2 试剂 硅胶G(青岛海洋化工厂);黄芩苷(批号110715-200212)、当归、桂枝等对照品、对照药材由药品生物制品检定所提供;甲醇为色谱纯,蒸馏水,其他所用试剂均为分析纯。
2 薄层定性鉴别
2.1 当归的TLC鉴别 取本品1g,研细,加乙醇20ml,加热回流30min,滤过,滤液浓缩至1ml,作为供试品溶液。将处方中不含当归的其他药材,按本品工艺及上述供试品制备方法,制成阴性对照溶液。取当归对照药材1g,加乙醇10ml,同上法制成对照药材溶液。照薄层色谱法(中国药典2005年版一部附录VIB)试验,吸取上述三种溶液各5μl,分别点于一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以正丁烷-醋酸乙脂(9:1)为展开剂,展开,取出,晾干,置紫外光灯365nm下检视。供试品色谱中,在与对照药材色谱相应位置上,显相同颜色的荧光斑点,阴性溶液无此斑点[2],见图1。
2.2 桂枝的TLC鉴别 取本品3g,研细,加石油醚(60~90℃)20ml,时时振摇,浸渍过夜,滤过,滤液挥散至约1ml,作为供试品溶液。将处方中不含桂枝的其他药材,按本品工艺制成样品,按上述供试品制备方法制成阴性溶液。取桂枝对照药材1g,同上法制成对照药材溶液。照薄层色谱法(中国药典2005年版一部附录VIB)试验,吸取上供试品溶液、对照药材溶液与阴性溶液各10μl,分别点于同一硅胶G薄层板上,以正已烷-醋酸乙脂(17:3)为展开剂,展开,取出,晾干,置紫外光灯365nm检视。喷以10%香草醛浓硫酸溶液,于105℃加热至斑点显色清晰,置日光下检视,供试品色谱中,在与对照药材色谱相应位置上,显相同颜色的斑点,阴性溶液无此斑点[3]。见图2、图3。
3 含量测定
3.1 色谱条件 色谱柱:ZorbaxEclipseXDBC18(150mm×4.6mm,5μm);柱温:室温。甲醇-水-磷酸溶液(40:60:0.2)为流动相,检测波长:280nm,流速:1.0ml/min。色谱柱理论塔板数按葛根素峰不低于2500[3]。
3.2 方法学考察
3.2.1 检测波长的选择 取黄芩苷加甲醇溶液于紫外分光光度计上绘制紫外吸收光谱,波长范围200~400nm,根据测定结果,最大吸收波长为280nm。
3.2.2 供试品制备 取本品10g,研细,精密称定0.3g,加70%甲醇40ml,加热回流3小时,放冷,过滤,滤液置100ml量瓶中,用少量70%甲醇分次洗涤容器壁和残渣,洗液滤入同一量瓶中,加70%甲醇至刻度,摇匀,精密吸取1ml,置10ml量瓶中,加甲醇至刻度,即得。
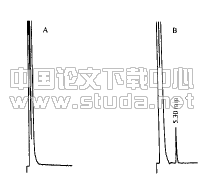
3.2.3 阴性试验 取除去黄芩以外的其它药材,按供试品制备方法制成。测定法,分别精密吸取供试品溶液、阴性液、对照品溶液各10μl,注入液相色谱仪中测定,绘制液相色谱图,由色谱图提示,在对照品色谱相应的位置上,供试品溶液具有相同保留时间的色谱峰,阴性液在此峰位无吸收,对本品中黄芩苷含量测定无干扰,方法专属性良好[3],见附图4、图5、图6。
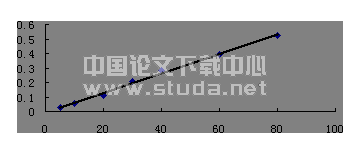
3.3 线性范围考察 精密称取黄芩苷对照品0.236mg/ml,分别精密吸取对照品溶液各1.0、2.5、5.0、7.5、10.0ml,置25ml量瓶中,加30%乙醇溶解并稀释至刻度,摇匀。分别精密吸取对照品溶液各10μl溶液,注入液相色谱器仪中,测定色谱峰面积,测定结果经线性回归,回归方程为:Y=3558.3X+36.098;r=0.9998,测定结果提示,黄芩苷进样量在0.0944~0.9440μg范围内线性关系良好。
3.4 稳定性试验考察 精密称取本品1.3g,按3.3.2项下制备供试液,精密取同一供试液,按0、2、4、6、8小时时间间隔,分别进样分析,测定色谱峰面积,测定结果RSD为1.40%,表明样品在8小时内基本稳
定。
3.5 精密度试验考察 精密称取本品1.3g,按3.3.2项下制备供试液,精密取同一供试液,分别精密吸取10μl,连续进样5次,按前述色谱条件进样分析,测定色谱峰面积,RSD为1.10%。
3.6 重复性试验考察 取同一批号样品,依法独立测定5次,结果RSD为1.12 %,表明本方法重现性良好。
3.7 回收率试验考察 精密称取已知含量样品(批号:20070108,含量12.57mg/g)5份,分别精密加入葛根素对照品溶液(16.01mg/ml)1ml,依前法测定,结果见表1。表1 回收率试验测定结果测定结果提示,本方法回收率在98.1~100.6%之间,RSD为1.50%,符合有关规定。
3.8 样品含量测定结果 经采用3.3.2项和3.2项下方法,测定供试品中黄芩苷含量,测定结果见表2。表2 3批样品含量测定结果(略)
4 讨论与结果
4.1 当归的薄层鉴别时,展距不宜过长,否则斑点容易扩散,且本色谱放置后斑点荧光逐渐减弱,应注意及时记录结果。
4.2 在含量测定中,我们针对流动相进行了筛选,本
实验先后采以甲醇-水-磷酸(47:53:0.2)(Ⅰ)[3],甲醇-水-磷酸(40:60:0.2)(Ⅱ),为流动相,在系统(Ⅱ)的条件下,供试溶液中黄芩苷与相邻组分分离度良好,阴性液无干扰。故确立甲醇-水-磷酸(40: 60:0.2)为流动相系统。
【】
[1] 张承烈.抗过敏中药研究进展[J].浙江中西医结合杂志,2005,15(4):34.
[2] 张军平.宫颈康颗粒质量标准研究[J].药房,2006,7(2):126.
[3] 药典委员会.中华人民共和国药典2005年版第一部[S].北京:化学出版社,2005:211.