脑脉通有效部位中总蒽醌的含量测定
作者:王淑美,冯素香,李淑芳,高淑娟,张丹,李建生
【摘要】 目的 建立脑脉通中大黄总蒽醌含量测定方法。方法 利用大黄蒽醌类化合物能与质量分数0.5%醋酸镁反应显色,在波长512 nm处产生吸收,采用可见分光光度法测定脑脉通复方中的以游离型蒽醌计的总蒽醌的含量。结果 以1,8?二羟基蒽醌为对照品,测得脑脉通有效部位中总蒽醌的含量在10%以上,平均回收率为102.91%,RSD=0.97%。结论 本方法简便、准确、重复性好,可作为脑脉通有效部位中大黄总蒽醌的含量测定方法。
【关键词】 比色法;脑脉通;总蒽醌
脑脉通由大黄﹑川芎﹑葛根等组成,具有解毒降浊﹑益气活血﹑涤痰开窍﹑泻下之功。该方脑梗死效果明显,可明显改善生活质量,对脑缺血再灌注损伤有明显的保护作用[1,2]。
我们采用大孔吸附树脂纯化工艺并结合药效学实验纯化脑脉通,并得到有效成分含量高,杂质含量少的有效部位[3,4]。根据复方有效部位及其主要化学成分的研究分析,确定脑脉通有效部位中的蒽醌类成分为其有效成分之一。目前总蒽醌的含量测定方法主要是比色法。本研究建立一个简便可靠的脑脉通总蒽醌的含量测定方法,可有效控制脑脉通有效部位的质量。
1 仪器与试药
仪器:METTLER?AE240型1/100000分析天平(瑞士梅特勒公司),RE52A型旋转蒸发仪(上海亚荣生化仪器厂),KQ?100型超声波清洗器(昆山市超声仪器有限公司),UV?2201SHIMADZU型分光光度计(日本岛津)。
材料:大黄、人参、葛根、川芎药材均购自河南郑州药材批发市场,经河南中医学院生药学科的陈随清教授鉴定分别为蓼科植物掌叶大黄Rheum palamatum L.的干燥根及根茎,五加科植物人参Radix ginseng C.A.Mey.的干燥根及根茎,豆科植物野葛Pueraria lobata(Wild.)Ohwi的干燥根,伞形科植物川芎Ligustic um chuanxiong Hort.干燥根茎。1,8?二羟基蒽醌对照品购自药品生物制品检定所。醋酸镁、甲醇等均为分析纯。
2 方法与结果
2.1 溶液的制备
2.1.1 对照品溶液的制备 精密称取1,8?二羟基蒽醌0.01009 g,置100 mL容量瓶中,加甲醇溶解并定容至刻度,摇匀即得。
2.1.2 供试品溶液的制备 精密称取有效部位样品10 mg至圆底烧瓶中,加冰醋酸?25%盐酸(体积比18∶2)20 mL[3],温水浴水解45 min,冷却,移入分液漏斗中,三氯甲烷萃取至三氯甲烷层近无色,弃去水层。合并三氯甲烷液,用蒸馏水洗至水层近无色,三氯甲烷层移入蒸发皿中水浴蒸干,甲醇定容至10 mL容量瓶中,即得。
2.1.3 阴性样品溶液的制备 按全方比例分别称取除大黄外其他药材适量,按有效部位制备工艺制得缺大黄的阴性样品,按“2.2”项方法制得阴性样品溶液。
2.2 检测波长的选择
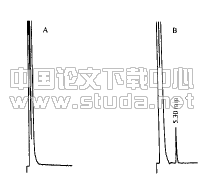
精密吸取对照品溶液2 mL,置25 mL容量瓶中,加质量分数0.5%醋酸镁甲醇溶液稀释至刻度,摇匀,即得。供试品溶液1 mL置10 mL的容量瓶中,加质量分数0.5%醋酸镁甲醇溶液稀释至刻度,摇匀,即得。按供试品显色方法制得阴性对照溶液。将上述溶液置于紫外分光光度计中,以质量分数0.5%醋酸镁甲醇溶液为空白参比溶液,400~800 nm范围内扫描测定吸收光谱。1,8?二羟基蒽醌对照品溶液加质量分数0.5%醋酸镁甲醇显色后的最大吸收波长为512 nm,供试品溶液在相同波长处有最大吸收,阴性供试品无干扰。见图1。
2.3 显色稳定性的考察
取显色后的对照品溶液和供试品溶液,分别放置0、0.5、1、2、4 h后,于512 nm处测定其吸光度。结果吸光度的RSD值分别为0.75%和0.47%,表明对照品和供试品溶液显色后4 h内基本稳定。
2.4 标准曲线制备
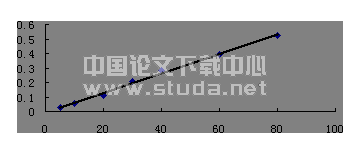
精密吸取对照品溶液(0.100 9 mg·mL-1) 0、0.5、1.0、2.0、3.0、4.0 mL置25 mL容量瓶中,用质量分数0.5%醋酸镁甲醇稀释至刻度,分别于512 nm处测定吸光度。以1,8?二羟基蒽醌对照品质量浓度为横坐标,吸光度为纵坐标,绘制标准曲线,并进行线性回归,得到回归方程:ρ=0.176+18.992A(r=0.999 2)。结果表明,1,8?二羟基蒽醌在2.018~16.144 μg·mL-1范围内与吸光度呈良好的线性关系。
A.1,8?二羟基蒽醌;B.供试品溶液; C.阴性样品
图1 紫外吸收光谱图(略)
Fig.1 UV spectrogram
2.5 精密度试验
精密吸取对照品溶液(0.100 9 mg·mL-1)2 mL置25 mL容量瓶中,用质量分数0.5%醋酸镁甲醇稀释至刻度,于512 nm处连续测定6次,记录其吸光度,结果表明,该方法的精密度良好。
2.6 稳定性试验
取有效部位样品10 mg,精密称定,按“2.2”项下方法制备供试品溶液,分别于配制溶液后0、1、2、3、4、6 h显色测定其吸光度,含量的RSD值为1.01%,表明样品溶液在6 h内稳定。
2.7 回收率试验
取同一批有效部位样品5份,每份约5 mg,精密称定,分别精密加入质量浓度为0.55 mg·mL-1的对照品溶液1 mL,蒸干后按“2.2”项下方法制备供试品溶液,最后定容至10 mL的容量瓶中。精密吸取供试品溶液1 mL置10 mL的容量瓶中,摇匀,即得。5份样品分别显色后于512 nm处测定吸光度,计算含量,总蒽醌的平均回收率为102.91%,RSD=0.97%。
2.8 重复性试验
取同一批有效部位样品5份,每份约10 mg,按“2.2”项下方法制备,分别显色后于512 nm处测定吸光度,计算含量,结果表明,该方法的重复性良好。
2.9 样品含量测定
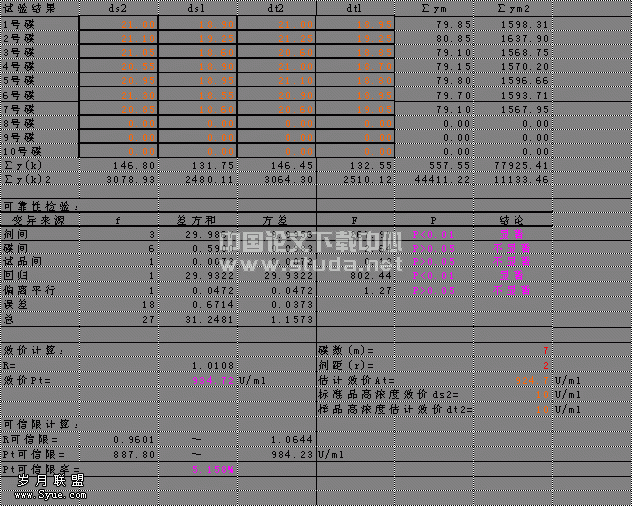
取3批有效部位样品各5份,每份约10 mg,精密称定,按“2.2”项下方法制备供试品溶液,最后定容至10 mL的容量瓶中。精密吸取供试品溶液1 mL置10 mL的容量瓶中,加质量分数0.5%醋酸镁甲醇溶液稀释至刻度,摇匀,即得。分别于512 nm处测定其吸光度,计算样品的含量。结果见表1。
3 讨论
3.1 《药典》2005年版一部中,大黄样品的制备采用的是8%的盐酸水解[5],而在实验过程中发现采用此方法测定含量有些偏低,因此我们对酸水解条件进行了筛选,选择了盐酸、硫酸、盐酸与冰醋酸的混合酸进行对比研究,由测定结果可知,采用盐酸与硫酸水解方法所测定的含量偏低,故选择盐酸与冰醋酸混合酸作为水解的方法。
表1 样品含量测定结果(略)
Tab.1 Results of samples determination(n=5)
3.2 本文建立了脑脉通有效部位中总蒽醌的含量测定方法,并进行了方法学考察试验研究,结果表明符合定量分析要求。该方法重复性、精密度、稳定性良好,而且操作简便,分析快速,可作为脑脉通有效部位质量控制方法之一。3批样品含量测定结果,固化物中总蒽醌的含量均高于10%。
【】
[1] 任小巧,李建生,封银曼,等.脑脉通对老龄大鼠脑缺血/再灌注损伤脑保护作用的研究[J].中国中药杂志,2004,29(1):66-68.
[2] 李建生,邓西方,任小巧.脑脉通对老龄大鼠脑缺血再灌注细胞因子的影响[J].中国医药学报,2004,19(8):471.
[3] 王淑美,冯素香,梁生旺,等.高效液相色谱法测定脑脉通有效部位中蒽醌类成分的含量[J].中国新药与临床杂志,2007,26(3):171-174.
[4] 王淑美,冯素香.脑脉通有效部位中总生物碱的含量测定[J].广东药学院学报,2008,24(1):1-3.
[5] 国家药典委员会.中华人民共和国药典:一部[S].北京:化学出版社,2005:17.