HPLC测定夏桑菊颗粒中蒙花苷的含量
作者:黄晓文,谢艳婷,冯嘉欣,谢銮凤
【摘要】 目的 建立夏桑菊颗粒中蒙花苷的测定方法。方法 采用反相高效液相色谱法,色谱柱:DiamonsilTMC18柱(250 mm×4.6 mm,5 μm,迪马公司);流动相为甲醇?四氢呋喃?水(43∶5∶52),检测波长334 nm,流速1.0 mL·min-1,柱温30 ℃。结果 蒙花苷在0.048~0.959 μg范围内线形关系良好,r=0.9999,平均加样回收率为100.7%,RSD为0.86%(n=6)。结论 所建立的方法准确,重现性好,灵敏度高,可用于该制剂的质量控制。
【关键词】 夏桑菊颗粒;蒙花苷;高效液相色谱法
夏桑菊颗粒收载于《卫生部药品标准》中药成方标准第15册,由夏枯草、野菊花、桑叶3味中药组成,具有清肝明目、疏风散热作用,用于风热感冒、目赤头痛、头晕耳鸣、咽喉肿痛等症。野菊花是本制剂的主要成分之一,蒙花苷是野菊花的特征成分[1-2],[3-5] 报道以高效液相色谱法测定野菊花制剂中蒙花苷含量,主要采用加甲醇超声处理或回流提取的方法,本文根据蒙花苷不溶于水的性质[6],以正丁醇萃取法提取蒙花苷,建立高效液相色谱法测定夏桑菊颗粒中蒙花苷含量的方法,结果准确、灵敏,阴性对照不干扰,为该制剂的质量控制提供依据。
1 仪器与试药
1.1 仪器
日本岛津高效液相色谱仪(LC?10Avp溶剂输送泵、SPD?10Avp紫外?可见检测器、SCL?10Avp系统控制器、CTO?10ASvp柱温箱);岛津Lcsolution工作站;日本岛津AUW220D天平;HH?2恒温水浴锅。
1.2 药品与试剂
蒙花苷对照品:由药品生物制品检定所提供,批号110752?200511,供含量测定用,质量分数95.9%。夏桑菊颗粒:广州星群(药业)股份有限公司。甲醇、四氢呋喃为色谱纯,水为双蒸水,正丁醇为分析纯。
2 方法与结果
2.1 溶液的制备
2.1.1 对照品溶液
精密称取经五氧化二磷干燥12 h的蒙花苷对照品5.00 mg,置100 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,得质量浓度为47.95 μg·mL-1的蒙花苷对照品贮备液。精密量取3.0 mL,置10 mL容量瓶,加甲醇稀释至刻度,摇匀,即得(质量浓度为14.39 μg·mL-1)。
2.1.2 供试品溶液
取装量差异项下的本品,研细,取5 g,精密称定,加水45 mL使溶解,用以水饱和的正丁醇提取 3次,每次正丁醇的用量分别为25、20、20 mL,合并正丁醇液;用以正丁醇饱和的水20 mL洗涤,洗涤后的水层用以水饱和的20 mL正丁醇提取,合并正丁醇液,水浴蒸干,残渣加甲醇适量使溶解,移至25 mL量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
2.1.3 阴性对照液
取处方中除野菊花外的其余成分制成不含野菊花的阴性对照品,按“2.1.2”项下的方法制备野菊花阴性对照溶液。
2.2 色谱条件及系统适应性试验
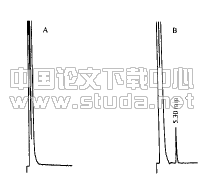
色谱柱:DiamonsilTM C18柱(250 mm×4.6 mm,5 μm,迪马公司);流动相∶甲醇?四氢呋喃?水(体积比43∶5∶52);流速:1.0 mL·min-1;检测波长334 nm;柱温为30 ℃。分别取蒙花苷对照品溶液、供试品溶液和野菊花阴性对照溶液各20 μL,按上述色谱条件,注入色谱仪。蒙花苷与样品中相邻色谱峰的分离度大于2.0,理论板数以蒙花苷应不低于2 500,阴性对照没有干扰。蒙花苷对照品、供试品溶液及阴性对照溶液的色谱图见图1、2、3。
2.3 方法学考察
2.3.1 线性关系考察
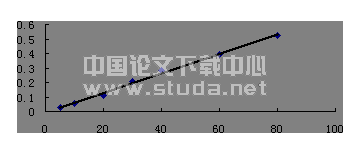
精密吸取0.5、1.0、2.0、4.0、6.0、8.0、10.0 mL的蒙花苷对照品贮备液,置10 mL的容量瓶中,用甲醇稀释至刻度,得质量浓度分别为2.40、4.80、9.59、19.18、28.77、38.36、47.95 μg· mL-1的系列对照品溶液。吸取上述系列对照品溶液各20 μL,分别进样测定,以峰面积(A)对溶液的质量浓度(ρ)进行回归,得回归方程A=43690ρ-5780,r=0.999 9,表明蒙花苷在0.048~0.959 μg之间与峰面积线性关系良好。
2.3.2 精密度试验
取蒙花苷对照溶液20 μL,注入色谱仪,连续测定6次,记录峰面积,结果蒙花苷峰面积的平均值为607 366,RSD为0.40%,表明仪器精密度良好。
2.3.3 重复性试验
取同一批号(批号DA11008)的样品6份,按“2.1.2”项下方法制备供试品溶液,进样测定,6次测定结果的RSD为1.44%,表明本方法重复性好。
2.3.4 稳定性试验
取同一供试品溶液(批号DA11008),分别在0、2、4、8、12、24 h进样20 μL,结果峰面积RSD为0.82%,表明供试品溶液在24 h内稳定。
2.3.5 加样回收试验
取已知含量(批号DA11008)的样品粉末约2.5 g,共6份,分别精密加入蒙花苷对照品贮备液(质量浓度为47.95 μg·mL-1)4.2 mL,按“2.1.2”项下方法制备溶液,按上述色谱条件下进样20 μL,测定,结果蒙花苷平均回收率为100.7%,RSD为0.86%,结果表明本方法准确可靠。结果见表1。表1 蒙花苷回收率试验结果(略)
2.4 样品测定
取夏桑菊颗粒样品3批,照“2.1.2”项下方法制备供试品溶液。取蒙花苷对照品溶液与供试品溶液各20 μL,注入色谱仪,测定,计算蒙花苷的含量。结果3批样品(批号DA11008、DA10788、DA10963)所测得的含量分别为0.929、0.897、0.706 mg.袋-1,RSD分别为0.5%、1.3%和1.1%(n=3)。
3 讨 论
3.1 蒙花苷提取方法的选择
分别考察了正丁醇萃取、甲醇回流、甲醇超声处理60 min等提取方法的提取效率,结果测得供试品(批号DA11008)中蒙花苷的含量分别为0.929、0.856、0.914 mg·袋-1,表明用正丁醇萃取时含量最高,且其他成分的干扰少, 蒙花苷与其他成分完全分离。 同时考察了正丁醇萃取的次数,分别将每次提取的正丁醇液蒸干后用甲醇定容至25 mL,进样测定,萃取1~4次制得的供试品溶液中蒙花苷的含量分别为353.4、52.9、8.6 μg和未检出;考察了以正丁醇提取以正丁醇饱和的水洗涤液的次数,结果提取1~2次的供试品溶液中蒙花苷的含量分别为3.5 μg和未检出, 故采用本方法制备供试品溶液。
3.2 关于检测波长的选择
采用SPD?M10A二极管阵列检测器,对蒙花苷对照溶液和供试品溶液,在200~600 nm波长处进行扫描,结果在波长为334 nm处有最大吸收,其他成分没有干扰,故选择334 nm作为检测波长。见图4、图5。
3.3 流动相的选择
曾[2-6]分别采用不同比例的甲醇?水、甲醇?水?磷酸、甲醇?水?冰醋酸、甲醇?0.1%磷酸、乙腈?0.5%磷酸作流动相,结果蒙花苷和本品中其他成分未能分离。经过对不同比例的甲醇?四氢呋喃?水进行试验,结果以甲醇?四氢呋喃?水(体积比43∶5∶52)为流动相时,蒙花苷可与其他组分完全分离,且峰形良好,该方法准确、灵敏,可用于本品的质量控制。
【参考文献】
[1]国家药典委员会.中华人民共和国药典:一部[S].北京:化学出版社,2005:219.
[2]谭晓杰,贾英,陈晓辉,等.RP?HPLC法测定野菊花中蒙花苷含量[J].沈阳药科大学学报,2004,21(6):434-435.
[3]张珊珊,彭艳梅,吴艳.HPLC法测定夏桑菊颗粒中蒙花苷含量研究[J].中医药导报,2007,13(4):79-80.
[4]卢丹,吕高荣,邹节明.HPLC测定玉叶解毒颗粒中蒙花苷含量[J].中药杂志,2005,30(3):191-192.
[5]陈在敏.HPLC法测定感冒灵颗粒中蒙花苷的含量[J].海峡药学,2005,17(4):50-51.
[6]陈晓辉,潭晓杰,田中克佳,等.制备型高效液相色谱在中药野菊花化学成分分离中的应用[J].药品评价,2004,1(5):359-361.