叉头蛋白、p66shc与长寿
来源:岁月联盟
时间:2010-07-12
【关键词】 叉头蛋白 p66shc DAF16 长寿
长生不老一直是人类的梦想,虽然发现许多因素能缩短寿命,但到目前为止,人们仍然没找到可供使用的延长寿命的办法。然而,近几年的遗传学研究发现敲除某些基因能延长线虫或小鼠的寿命,这给长寿的研究带来新的希望。
1 p66shc和长寿
哺乳类的原癌基因shc编码三个分子量相近的蛋白:52K(P52shc)、46K(p46shc)和66K(p66shc)。它们都有src同源结构域(SH2),一个胶原-同源区(CH1)和磷酸酪氨酸结合结构域(PTB)。仅p66shc含有氨基末端区(CH2)(图1)。
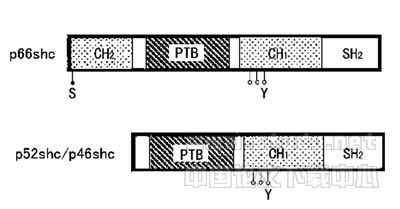
图1 p66shc、p52shc和p46shcde结构图解
S:S36,P66shc的主要丝氨酸磷酸化部位;Y:酪氨酸磷酸化部位
为研究p66shc在应激反应上的作用,Migliaccio等[1]使用测定p66shc磷酸化敏感的phospho-qmino-acid分析方法的研究证明,用UV和H2O2 处理细胞后磷酸丝氨酸明显而持续地增加,为研究p66shc 在应激反应上的作用,Migliaccio等研究了敲除p66shc在细胞对氧化和DNA损伤(UV和γ射线照射)反应上的作用。MEFs细胞是来自有shc CH2定点突变的小鼠,这种突变不影响p52shc和p46shc的编码序列。像期望的那样,p66shc在 p66shc+/-细胞是降低的,在 p66shc-/-细胞是缺乏的,但在这种细胞中,p52shc和p46shc都是正常的。野生型MEFs对H2O2 处理是敏感的,即在处理24h后70%细胞死亡(其中的70%以上是凋亡),超表达p66shc 增加野生MEFs的易感性(80%~90%的细胞死亡)。而 p66shc的敲除增加MEFs对H2O2 的抵抗(<30%的细胞死亡)。如用UV处理细胞,在野生型,处理24h和48h以后,分别有80%和40%的细胞仍然生存,在p66shc-/-,UV对细胞未显示出毒性作用。研究还证明,仅野生型p66shc在 p66shc-/-细胞中的表达即可恢复细胞对UV和H2O2 的正常反应,但p52shc和p46shc的表达没有类似的作用。
为检查p66shc-/-在体内抵抗氧化应激的能力,动物用能产生超氧阴离子的百草枯(paraquat)处理,10周大的小鼠用70mg/kg的百草枯腹腔注射,5只野生型小鼠在注射48h内全部死亡,但5只p66shc-/- 小鼠在48h内仅死亡2只,另2只在72h后死亡,一只存活了7周。研究还发现p66shc能影响寿命,一个研究使用了36只小鼠,其中野生型14只,8只杂合子(p66shc+/-),14只的p66shc的被敲除(p66shc-/-)。这些小鼠都是p66shc+/-杂合子双亲所生,在同样的条件下饲养。28个月以后,野生型小鼠全部死亡,8只杂合子中的5只死亡,而14只p66shc敲除小鼠仅死亡3只。
2 p66shc和叉头蛋白的机能联系
叉头蛋白转录因子家族成员包括DAF-16和哺乳类的FKHRL1、FKHR和AFX等。它们都有一个高度保守的100个氨基酸的叉头序列,这个序列含有一个有翼螺旋(winged helix)并具有DNA结合活性2]。遗传学分析证明叉头蛋白转录因子家族成员在细胞分化和增生上起重要作用[3],晚近的研究还表明它们和肿瘤的发生密切相关[4]。这些转录因子可以被蛋白激酶B(也称为Akt)磷酸化,磷酸化可以抑制其转录活性[5]。
为探讨FKHRL1转录活性和细胞内活性氧(ROS)的关系,Nemoto等[6]研究了H2O2处理和FKHRL1磷酸化的关系,他们发现H2O2处理培养的PC12细胞导致浓度依赖的FKHRL1磷酸化的增加。他们还发现可溶性抗氧化剂谷胱甘肽前体-已酰基半胱氨酸和谷胱甘肽都能以浓度依赖的方式降低PC12细胞由H2O2处理诱发的FKHRL1的磷酸化。
到目前为止,能增加哺乳类动物寿命的遗传改变是敲除p66shc的纯合子,p66shc能调节细胞内的ROS水平,并因此可以修饰叉头蛋白的活性。研究证明虽然小鼠的野生型(p66shc+/+和p66shc-/-型的成纤维细胞中的H2O2水平都很低,细胞在受到可增加氧化应激的血清撤除刺激以后,同野生型相比,p66shc-/-细胞的H2O2水平更低。
P66shc的36位丝氨酸的残基的突变将产生一个显性干扰表型(dominant interfering phenotype),仅用空载体、野生型p66shc+/+和有36位丝氨酸突变的p66shc分别转染PC12细胞,虽然转染野生型p66shc和有36位丝氨酸突变的p66shc的PC12细胞超表达的p66shc蛋白的量是相似的,但表达有36位丝氨酸突变的p66shc的PC12细胞中的ROS的水平比表达野生型的细胞低,它的量近似于p66shc-/-细胞。
对有延长寿命突变的线虫的遗传学研究也证明[7],适当增加叉头蛋白的活性能延长寿命。研究者发现p66shc-/-细胞中的依赖叉头蛋白的转录活性是增加的,它在导致强烈氧化应激反应的血清撤除时更明显。还有报道证明p66shc显性干扰突变的稳定表达也可增加FKHRL1的活性。为证明氧化还原依赖的叉头蛋白的失活能被p66shc调节,细胞被用外源性H2O2处理,野生型成纤维细胞在H2O2 处理以后,FKHRL1的磷酸化快速和有统计学意义的增加,而p66shc-/-细胞在氧化应激处理后FKHRL1的磷酸化无明显改变,在不表达p66shc的细胞,同时p66shc-/-细胞在接受其他应激 (如紫外线) 刺激以后,FKHRL1的磷酸化也无明显改变。在表达突变的p66shc 的PC12细胞也观察到类似的FKHRL1的调节模式。在p66shc-/-成纤维细胞中仅表达野生型p66shc-/-就能恢复氧化应激诱发的FKHRL1磷酸化,这表明氧化应激修饰的插头蛋白磷酸化需要p66shc的参与。在线虫,DAF-16之所以能调节氧化应激,部分是由于它直接或间接反式激活(transactivation)许多抗氧化酶和一些应激基因相关产物。为探讨在哺乳类细胞增加FKHRL1的转录活性是否可提高抗氧化清除和对氧化应激的抵抗能力,Nemoto等分析了FKHRL1修饰的过氧化氢酶启动子的反式激活,他们识别了大量从人的过氧化氢酶启动子的-2339和-1667部位开始的FKHRL1的共有结合序列。野生型的FKHRL1可反式激活含有3kb的过氧化氢酶启动子片断的报告子构件,但不能反式激活缺少共有结合部位的较短的1.6kb片断的报告子构件。3kb的过氧化氢酶启动子片断的转录活性在大小上类似于含有前后排列的FKHRL1结合部位的合成报告子的转录活性。像期望的那样,当用有活性结构域丢失突变的FKHRL1转染细胞时,无论全长的过氧化氢酶启动子还是合成的叉头蛋白启动子的转录活性都不增加。此外,仅超表达野生型FKHRL1的PC12细胞表现出过氧化氢清除能力的提高并在过氧化氢处理后提高生存能力。
叉头蛋白转录因子DAF-16是胰岛素/胰岛素样生长因子信号传导通路的下游效应器,在蠕虫、果蝇和小鼠胰岛素/胰岛素样生长因子信号传导通路能使DAF-16失活。最近的研究结果表明[8],DAF-16在果蝇成体脂肪体中超表达能延长其寿命和降低生育频率,果蝇的脂肪体相当于哺乳类的肝和脂肪组织,而降低胰岛素/胰岛素样生长因子信号传导通路的活性,同样能延长蠕虫、果蝇和小鼠的寿命。
总之,这些研究表明叉头蛋白、p66shc和细胞内ROS之间存在重要的机能联系,其中叉头蛋白在线虫、果蝇和小鼠可调节寿命的长短,而ROS在所有种属的老化上起重要作用[9,10]。
【】
1 Migliaccio E. Nature,1999,402:309-313.
2 Biggs WH. Proc Natl Acad Sci USA,1999,96:7421-7426.
3 Romas R, Costa R. Crit Rev Oncol Hematol,1995,20: 129-140.
4 Vogt PK. Virology,1997,283:1-7.
5 Brownawell AM. Mol cell Biology,2001,21: 3534-3546.
6 Nemoto S,Finkel T. Science,2002,295: 2450-2452.
7 Taub J.Nature,1999,399: 162-166.
8 Leevers J,Partridge L. Science,2004,305:361.
9 Finkel T,Holbrook NJ. Nature,2000,408: 239-247.
10 Biggs WH.Proc Natl Acad Sci USA,1999,96:7421-7426.











